當團隊開發一套新工具,要如何評估工具的準確度?這時需要一組可信度高的正確資料集來協助衡量(benhcmark)工具表現,以下介紹在人類基因體常用的資料集來源。
GIAB
Genome In A Bottle Consotrium 由美國國家標準與技術研究所 (NIST) 主持的計畫,主要提供人類基因體相關的標準品與正確資料集(truth set):
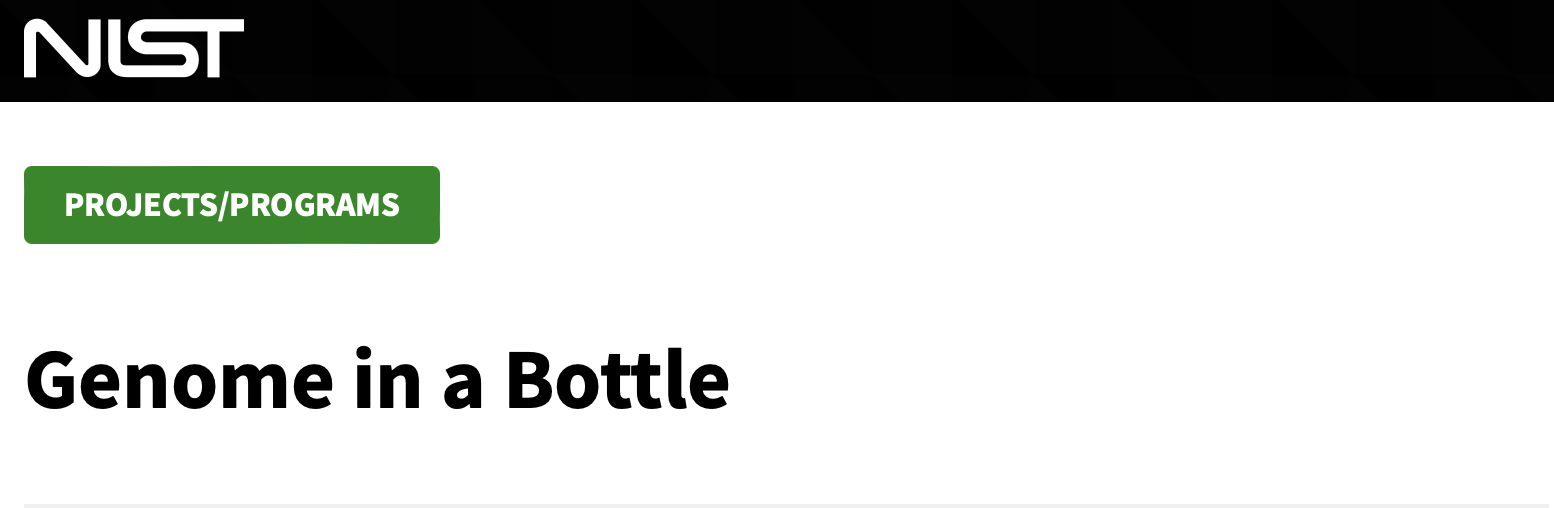
標準品主要從NIGMS Human Genetic Cell Repository挑選cell line 來產生,共有7個樣本(HG001-HG007),標準品的編號和族群、家族關係等整理如下表:
(table collected from Coriell Institute for medical research)
製作正確資料集首先需要不同定序平台與偵測工具的檔案來源、在生資分析上也需要做好品質篩選(QC)、變異的比對合併(merging)以及基因型計算(genotyping)過程可說耗費精力。GIAB不僅提供多個標準品的正確資料集,也致力於提供不同種變異類型/參考序列的資料集,目前官網有提到以下幾種:
Structural variants: Currently available for HG002 on GRCh37 and in Challenging Medically Relevant Gene benchmark below
Small variants in more difficult regions: v4.2.1 is available for all 7 GIAB samples on GRCh37 and GRCh38 (manuscript).
MHC: Included in v4.2.1 small variant benchmark for HG001-HG007 (Manuscript describing MHC benchmark)
273 Challenging Medically Relevant Genes small variant and SV benchmarks in HG002 and Preliminary benchmark for T2T-CHM13v1.0
v1.0 TR benchmark for HG002 indels and SVs >=5bp in tandem repeats on GRCh38 (preprint)
v1.0 XY benchmark for HG002 small variants in chromosomes X and Y on GRCh38 (preprint)
GIAB團隊以令人驚訝的速度前進,久沒關注竟然有針對tandem repeat跟性染色體的truth set,目前僅先釋出資料集給研究者,論文似乎還在preprint階段
資料下載
如果想要truth set,可以直接從ftp網站找檔案下載使用: https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/
或是去 GitHub 查看感興趣的標準品目前有沒有對應的 truth set: https://github.com/genome-in-a-bottle/giab_latest_release
兩個路徑都行,反正最後都會引導到相同的地方
實際找個HG002 SNV/indel GRCh38 truth set來看,透過層層資料夾點選,最終找到目標VCF檔:

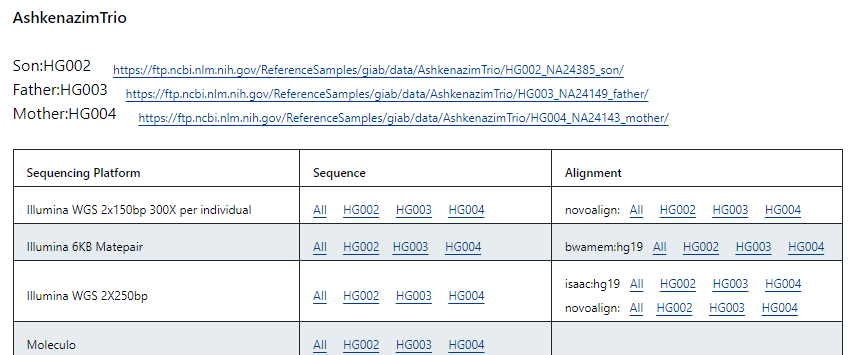
如果想要看開源的標準品fastq, bam 等檔案,以拿來當作測試工具的input,可以至以下網站查詢: https://github.com/genome-in-a-bottle/giab_data_indexes
網站也是先依族群分標準品,然後分定序平台和檔案類型:
Somatic truth set
上述可能針對human germline variant 為主,而想做somatic caller benchmarking 的話該如何?最近找到一個Biostar的討論,裡面有提到兩篇釋出somatic truth set的文章,一個是WGS, 一個針對panel,最近準備來讀:
Fang, L.T., Zhu, B., Zhao, Y. et al. Establishing community reference samples, data and call sets for benchmarking cancer mutation detection using whole-genome sequencing. Nat Biotechnol 39, 1151–1160 (2021). https://doi.org/10.1038/s41587-021-00993-6
Jones, W., Gong, B., Novoradovskaya, N. et al. A verified genomic reference sample for assessing performance of cancer panels detecting small variants of low allele frequency. Genome Biol 22, 111 (2021). https://doi.org/10.1186/s13059-021-02316-z
討論
過去在沒有資料及整合釋出前,會使用模擬資訊來作為正確答案;之後可能會使用同時具有WGS, WES等不同定序結果的樣本,透過不同層級的證據來驗證變異真實存在,或是使用sanger sequencing等實驗做驗證,但缺點為無法一次比較出結果,且驗證樣本的品質等因素也會影響評估結果。
若對truth set 流變和其他團隊製作的truth set感興趣的話,可閱讀2023年這篇 review paper:
Majidian, S., Agustinho, D.P., Chin, CS. et al. Genomic variant benchmark: if you cannot measure it, you cannot improve it. Genome Biol 24, 221 (2023). https://doi.org/10.1186/s13059-023-03061-1
文章的corresonding author Fritz J. Sedlazeck 和 Medhat Mahmoud 都是 GIAB 的團隊領導,也參與美國許多定序計畫如All of US, T2T consortium 等
不過雖然說是truth set,其製作過程也是將不同平台與工具偵測的結果經由比對,篩選留下的consensus cell-set,若未來隨著定序技術進步,如PacBio, Nanopore等long-read定序資料占比提高,一定會有更多位點被偵測到,或是篩掉某些位點;另外不同參考序列的取捨,也會影響留在truth set的位點資訊,如從GRCh37 truth set 經由座標轉換(liftover)至 GRCh38 後,拿來benchmark GRCh38 detected test set 就會有一些潛在無法比對的區域,導致false positive 數量增加。
而有正確解答和代測檔案,就可以找做benchmarking的工具了,如 snv/indel比對的hap.py等,有機會可以寫一篇來介紹。